

In this article, I briefly describe how a primary deficiency leads to different immunodeficiency syndromes.
Primary Immunodeficiency
Primary immunodeficiency is generally a birth defect and causes many health issues in early life. Immunodeficiency disorders are minor or life-threatening. Primary immunodeficiency diseases have provided immunologists valuable insights into the critical roles of specific proteins and cellular processes necessary for a well-functioning immune system. These conditions range in severity from mild to life-threatening if left untreated or if infections are not adequately controlled. They are generally classified based on whether they impact innate or adaptive immunity and are often further categorized by the particular immune system components most affected, including B cells, T cells, combined B and T cells, phagocytes, innate immunity, and complement systems.
Defects in the Major Histocompatibility Complex (MHC) Cause Bare-Lymphocyte Syndrome
If MHC class I or class II molecules are not expressed in an individual, it leads to general failures of immunity. Positive selection of CD4+ T cells in the thymus is impaired in the absence of class-II MHC. Peripheral T helper cells show a limited response. Deficiencies in MHC class I or class II result in bare-lymphocyte syndrome. This syndrome is a rare disorder caused by mutations in β2 microglobulin, a subunit of class-I MHC protein. The TAP proteins play a vital role during antigen presentation by MHC class I molecules. Mutations in TAP proteins also lead to immunodeficiency disorders. The affected MHC class I proteins result in impaired positive selection of CD8+ T cells, depressed cell-mediated immunity, and an elevated susceptibility to viral infection.
DiGeorge Syndrome (DGS) causes an Undeveloped Thymus
Some immunodeficiency syndromes affect T cells and eventually result in an undeveloped thymus. DiGeorge syndrome (DGS) is the outcome of some deletions in a region on chromosome 22, containing up to 50 genes. The T-box transcription factor (TBX1) present on chromosome 22 is highly expressed during certain specific stages of embryonic development, such as during the formation of facial structures, heart, thyroid, parathyroid, and thymus tissues.
Individuals suffering from DiGeorge syndrome have symptoms of abnormal facial features, hypoparathyroidism, and congenital heart anomalies. Patients suffering from complete DGS are devoid of thymic tissue and have a very small number of T cells with poor antibody responses. Many opportunistic pathogens can easily pounce on these patients. Passive antibody treatment and thymic transplantation can be taken as treatment options. Though the immune defects get corrected, severe heart disease can thwart the long-term survival of a patient.
A Mutation in an X-linked Gene Results in Wiskott-Aldrich Syndrome
Patients suffering from Wiskott-Aldrich syndrome (WAS) have a mutation in an X-linked gene, WASP. This gene encodes a cytoskeletal protein expressed highly in hematopoietic cells. The WAS protein assembles and reorganizes actin filaments in cells of the hematopoietic lineage. This assembly of actin filaments is important in proper synapse formation and intracellular signaling.
The clinical manifestations vary widely and generally appear early in the first year of life. The syndrome severity depends on the specific mutation. However, low platelet count (thrombocytopenia) and eczema are common among patients. Individuals with this syndrome often have humoral immune defects, affected cell-mediated immunity, and below-normal levels of IgM antibody. Patients frequently acquire bacterial infections, especially by encapsulated strains such as Streptococcus pneumoniae, Haemophilus influenzae type b (Hib), and Staphylococcus aureus. Along with the progression of the disease, the function of regulatory T cells is impaired, which results in autoimmunity and B-cell malignancy. Severe forms of the disease need hematopoietic stem cell transfer. Whereas mild cases of the disease can be managed by addressing the symptoms. This includes using transfusions to control bleeding and administering passive antibodies or antibiotics for bacterial infections.
Mutations in various proteins involved in signaling pathways downstream of TCRs can negatively impact T-cell development, peripheral T-cell activation, and essential functions like cell-mediated cytotoxicity. These include defects in the protein kinases Lck, ZAP-70, ITK, and other proteins necessary for cytoskeletal reorganization. Additionally, antibody responses may be impaired, and immune dysregulation can lead to autoimmunity.
Defects in CD40 or CD40L cause hyper-IgM Syndrome
B cells and other antigen-presenting cells, such as dendritic cells, monocytes, and macrophages, express CD40. T cells express CD40 ligand (CD40L). An inherited deficiency in either CD40 or CD40L leads to obstructed interaction between T cells and antigen-presenting cells. CD40L deficiency is an X-linked disorder, and CD40 deficiency is less common, controlled by an autosomal gene.
These deficiencies hinder the normal interaction between CD40L on TH cells and CD40L on B cells. This co-stimulatory interaction between CD40 and CD40L activates B cells through T helper cells. In the absence of this co-stimulation, B cells can’t mount normal responses to T-dependent antigens. The formation of germinal centers, somatic hypermutation, the heavy-chain class switching necessary for the production of IgG, IgA, and IgE antibodies, and the production of memory cells are hindered (Figure 1). However, B-cell responses to T-independent antigens remain unaffected and result in the rise of IgM antibodies. Patients with abnormally high IgM antibodies suffer from hyper-IgM syndrome.
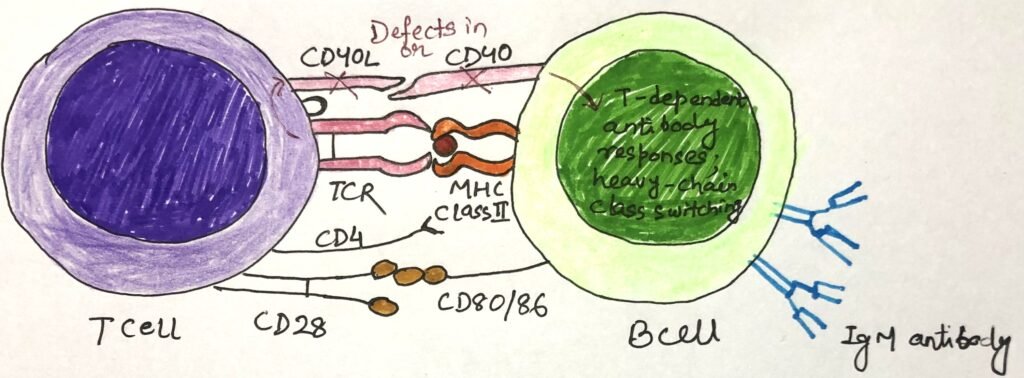
The TH activation of dendritic cell maturation and IL-12 secretion depends upon the CD40L-CD40 interaction. Any defects in this pathway result in no cell-mediated immunity and increased susceptibility to intracellular pathogens. Children with CD40L or CD40 defects suffer from different viral and fungal infections.
Elevated levels of IgE cause hyper-IgE Syndrome
Hyper IgE syndrome, also known as Job syndrome, is caused by an autosomal dominant mutation in the STAT3 gene. It is a primary immunodeficiency characterized by skin abscesses, recurrent pneumonia, eczema, and heightened IgE antibodies. The symptoms are associated with facial abnormalities and bone fragility.
The cytokines IL-6, IL-10, and IL-21 activate an intracellular signaling cascade, where the STAT3 gene is also involved. This signaling is important for TH17 and TFH cell differentiation. In the absence of the STAT3 gene, induction of IL-17, IL-10, IL-22, and TGF-β is hindered in response to antigenic stimulation.
The patients having Job syndrome have lower than normal levels of circulating TH17 cells. TH17 response is important for the clearance of fungal and extracellular bacterial infections. Thus, patients with Job syndrome have low TH17 responses and are susceptible to fungal and bacterial infections. In these patients, lower numbers of antigen-specific memory B cells cause reduced TFH cell activity. Reduced activity of TFH cells causes lower numbers of antigen-specific memory B cells. Patients having defects in the STAT3 gene also have hindered IL-10 signaling with undeveloped TREG cells. The involvement of STAT3 in the signal transduction of many cytokines could elevate the level of IgE antibodies. However, individuals with hyper-IgE syndrome do not show abnormal levels of allergic responses.
Conclusion
Primary immunodeficiency is generally a birth defect and causes many health issues in early life. Immunodeficiency disorders are minor or life-threatening.
If MHC class I or class II molecules are not expressed in an individual, it leads to general failures of immunity. Positive selection of CD4+ T cells in the thymus is impaired in the absence of class-II MHC. Deficiencies in MHC class I or class II result in bare-lymphocyte syndrome.
Some immunodeficiency syndromes affect T cells and eventually result in an undeveloped thymus. DiGeorge syndrome (DGS) is the outcome of some deletions in a region on chromosome 22, containing up to 50 genes. Patients suffering from complete DGS are devoid of thymic tissue and have a very small number of T cells with poor antibody responses. The syndrome severity depends on the specific mutation. However, low platelet count (thrombocytopenia) and eczema are common among patients.
B cells and other antigen-presenting cells, such as dendritic cells, monocytes, and macrophages, express CD40. T cells express CD40 ligand (CD40L). An inherited deficiency in either CD40 or CD40L leads to obstructed interaction between T cells and antigen-presenting cells. Defects in CD40 or CD40L cause hyper-IgM syndrome.
Hyper IgE syndrome, also known as Job syndrome, is caused by an autosomal dominant mutation in the STAT3 gene. It is a primary immunodeficiency characterized by skin abscesses, recurrent pneumonia, eczema, and heightened IgE antibodies.
You may also like:
- Primary immunodeficiencies are mostly hereditary
- Severe combined immunodeficiency (SCID) interrupts with T-cell and B-cell responses
- Defects in innate immune components result in deficiency and diseases
- Immunodeficiency disorders can lead to the development of autoimmunity

I, Swagatika Sahu (author of this website), have done my master’s in Biotechnology. I have around fourteen years of experience in writing and believe that writing is a great way to share knowledge. I hope the articles on the website will help users in enhancing their intellect in Biotechnology.