
In this article, I briefly describe the enzyme-linked immunosorbent assay, which is an antigen-antibody interaction.
Antigen-antibody interaction
The Interaction between antigen and antibody is a bimolecular association, which does not lead to an irreversible chemical alteration in either the antibody or the antigen. The antigen-antibody association involves many non-covalent interactions between the antigenic determinant (epitope) of the antigen and the variable-region (VH/VL) domain of the antibody molecule. The antigen-antibody binding depends on weak and non-covalent interactions like hydrogen bonds, hydrophobic interactions, electrostatic forces, and Van der Walls interactions. Thus, to make a sturdy antigen-antibody interaction, a large number of such weak interactions are required. These interactions can only take place if the antigen and antibody molecules are close enough for some of the individual atoms to fit into complementary recesses.
Affinity and avidity
A very close fit between antigen and antibody increases the strength of the bond. The sum of the attractive and repulsive forces operating between the antigenic determinant and the combining site of the antibody determines the affinity. The affinity of an antibody for a specific epitope is the combined strength of the non-covalent interactions between a single antigen-binding site on an antibody and the epitope.
The strength of multiple interactions between a multivalent antibody and antigen is called avidity. When complex antigens containing multiple repeating antigenic determinants are mixed up with antibodies containing multiple binding sites, the interaction of an antibody with an antigen at one site will increase the probability of a reaction between those two molecules at a second site. Avidity is more than the sum of the individual affinities. Affinity defines the strength of interaction between antibody and antigen at single antigenic sites, whereas avidity defines the overall stability or strength of the antibody-antigen complex. The strength of the antibody-antigen complex is controlled by three major factors, i.e., antibody-epitope affinity, the valence of both the antigen and antibody and the structural arrangement of the interacting parts.
Specificity and cross-reactivity
The specificity of an antigen-antibody reaction is the ability of an individual antibody combining site to react with only one antigenic determinant. It also defines the ability of a population of antibody molecules to react with only one antigen. An antibody can interact with its antigen, thus making the antigen-antibody reactions highly specific. A strong antigen-antibody interaction depends on a very close fit between the antigen and antibody, which requires a high degree of specificity.
Sometimes, the antibody elicited by one antigen can cross-react with an unrelated antigen, called cross-reactivity. The cross-reacting antigen has an epitope, which is structurally similar to one on the immunizing antigen.
Types of antigen-antibody interaction
There are mainly six types of antigen-antibody interaction and can be categorized as
- Precipitation reaction
- Agglutination reaction
- Complement fixation
- Immunofluorescence
- ELISA- Enzyme linked immunosorbent assay
- Radioimmunoassay (RIA)
Enzyme-linked immunosorbent assay (ELISA)
The ELISA is an immunological assay first described by Eva Engvall and Peter Perlmann in 1971. It uses a solid-phase enzyme immunoassay (EIA) to measure antibodies, antigens, proteins, and glycoproteins in biological samples. It has been used as a diagnostic tool in medicine and plant pathology, as well as a quality-control check in various industries. This test is based on an enzyme-substrate reaction that generates a colored reaction product. An enzyme conjugated with an antibody reacts with a colorless substrate to generate a colored reaction product. Various enzymes, such as alkaline phosphatase, horseradish peroxidase, and β-galactosidase, have been used for ELISA.
Qualitative detection or quantitative measurement of either antigen or antibody can be done by several variations of ELISA. There are four types of ELISA methods, e.g., direct ELISA, indirect ELISA, sandwich ELISA, and competitive ELISA. Antibody can be determined with an indirect ELISA whereas antigen can be determined with a sandwich ELISA or competitive ELISA. Each assay can be used qualitatively or quantitatively by comparison with standard curves prepared with known concentrations of antibodies or antigens.
Direct ELISA
It is the simplest type of ELISA. This method is used to detect and measure antigen concentration in a sample. In direct ELISA, the presence of a particular antigen in a sample is detected by using a capture monoclonal antibody. The procedure for direct ELISA is given below.
Procedure
- A microtiter plate is taken, and the wells of the plate are coated with a sample containing the target antigen. The antigen is fixed to the surface.
- The wells of the microtiter plate are then coated with a blocking buffer.
- In another reaction, an enzyme is linked to an antibody.
- In the next step, the enzyme-antibody conjugate is added to the wells to adsorb to the antigen.
- Then, the plate is washed to remove any excess enzyme-antibody conjugate.
- In the next step, a substrate is added for the enzyme, and the enzyme converts it into a product, which elicits a chromogenic or fluorescent signal. So, the substrate detects the presence of the enzyme and the antigen.
- The amount of colored product is measured by a specialized spectrophotometric plate reader.
The direct ELISA is relatively quick because of the use of only one antibody. However, it needs the labeling of every primary antibody, is time-consuming and more expensive than indirect methods. Certain antibodies may not be suited for direct labeling. Direct methods do not allow for signal amplification in contrast to methods that use a secondary antibody. The method can be used to test specific antibody-to-antigen reactions and helps to eliminate cross-reactivity between other antibodies.
Indirect ELISA
Among the four different types of ELISA, the method of indirect ELISA is applied for the detection of antibodies. It is the method of choice to detect the presence of serum antibodies against human immunodeficiency virus (HIV), the causative agent of AIDS. The procedure for indirect ELISA is given below.
Procedure
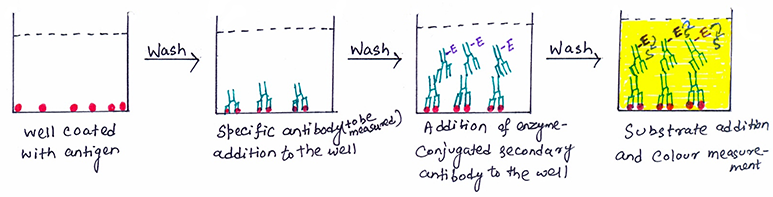
- A buffered solution of the antigen is added to each well of a microtiter plate. Antigen coating takes place through passive adsorption of the antigen to the assay microplate. This process involves the hydrophobic interactions between the microtiter plate and non-polar protein residues.
- After incubation, the wells are washed with neutral phosphate-buffered saline (PBS) or deionized water to remove any excess antigen. To block any plastic surface in the well that remains uncoated by the antigen, a solution of non-reacting protein, such as bovine serum albumin or casein, is added.
- Serum or some other sample containing primary antibody is added to the antigen-coated microtiter well. The antibody specific to the test antigen binds with the coated antigen on incubation.
- Any excess or unbound antibodies are washed and then added with a blocking solution.
- An enzyme-conjugated secondary anti-isotype antibody is added to detect the presence of bound antibody to the antigen. The enzyme-conjugated secondary antibody binds to the primary antibody. (figure 1).
- Any free secondary antibody is washed away, and a substrate for the enzyme is added. The color change of the substrate on reacting with the enzyme shows the binding of the secondary antibody to the primary antibody. The stronger change in color of the substrate shows the higher concentration of the primary antibody in the serum. The amount of colored reaction product that forms is measured by specialized spectrophotometric plate readers.
- The process of indirect ELISA helps to determine serum antibodies to HIV within six weeks of infection. In this assay, recombinant envelope and core proteins of HIV are adsorbed to solid-phase antigens to microtiter wells. Individuals infected with HIV will produce serum antibodies to epitopes on these viral proteins.
Advantages and disadvantages
Indirect ELISA has sensitivity as more than one labeled antibody is bound per primary antibody. It is also flexible since different primary detection antibodies can be used with a single labeled secondary antibody. Along with sensitivity and flexibility, indirect ELISA is cost-effective, as it requires fewer labeled antibodies.
However, Indirect ELISA has some disadvantages too. One of the major disadvantages of the process is the non-specificity of antigen immobilization. When serum is used as the source of test antigen, all proteins in the sample may stick to the microtiter plate well. So, small concentrations of analyte in serum must compete with other serum proteins when binding to the well surface. The sandwich or direct ELISA provides a solution to this problem by using a “capture” antibody specific for the test antigen to pull it out of the serum’s molecular mixture.
Sandwich ELISA
It measures the antigen between two layers of antibodies, thus also known as sandwich ELISA. The two layers of antibody consist of capture and detection antibody. In this process, either monoclonal antibodies or polyclonal antibodies can be used as capture and detection antibodies. The antigen to be measured must contain at least two antigenic sites capable of binding to antibody since at least two antibodies act in the sandwich. The procedure for sandwich ELISA is given below.
Procedure
- In sandwich ELISA, the capture antibody (capture antibody) is coated on the surface of a microtiter well.
- Any non-specific binding sites on the surface are blocked with the help of a blocking solution.
- The sample containing antigen is added to the well. It reacts with the immobilized antibody on the microtiter well.
- The microtiter plate is washed to remove any unbound or excess antigens.
- After washing the microtiter plate, a second enzyme-linked antibody, also known as a detection antibody specific for a different epitope on the antigen, is added. It reacts with the bound antigen.
- Then, the plate is washed to remove the unbound antibody-enzyme conjugates.
- Any free second antibody is washed, and the substrate is added. The colored reaction product is measured (figure 2). TMB (3, 3’, 5, 5’-tetramethyl benzidine) is the most commonly used substrate for the enzyme horseradish peroxidase (HRP).
- To determine the presence and quantity of antigen, the absorbency of the plate wells is measured. Specially designed spectrophotometers read through the microtiter wells either singly or in rows.

Advantage
In sandwich ELISA, the sample does not have to be purified before analysis. The assay can be very sensitive when compared to indirect ELISA or competitive ELISA. The two antibodies help in capturing and detecting the antigen, thus giving the process high specificity. The process has both flexibility and sensitivity.
Competitive ELISA
It is also known as inhibition ELISA and measures the concentration of antigen by detection of signal interference. The procedure for competitive ELISA is given below.
Procedure
- In the first step, an unlabeled primary antibody is incubated in solution with a sample containing antigen.
- The reference antigen is pre-coated on a microtiter well.
- Then, the antigen-antibody mixture is added to the reference antigen-coated microtiter well (figure 3).
- The more the antigen in the sample, the less free antibody will be available to the reference antigen-coated well. This leads to the development of competition.
- The plate is washed to remove any unbound antibodies.
- The enzyme-conjugated secondary antibody specific for the isotype of the primary antibody is added to determine the amount of primary antibody bound to the well.
- In the final step, a substrate is added, and the color change is measured.
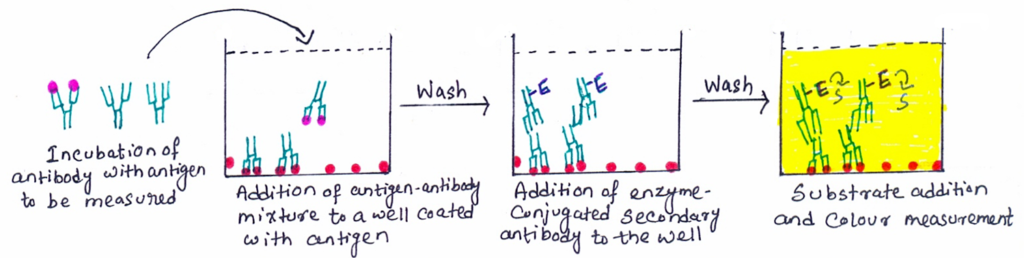
In competitive ELISA, the concentration of antigen in the original sample is inversely proportional to the color produced. Some competitive ELISA kits include enzyme-linked antigens rather than enzyme-linked antibodies. So, in this case, the microtiter well is coated with an antibody. The labeled antigen competes for primary antibody binding sites with the sample antigen (unlabeled). The more antigens present in the sample, the less labeled antigen is retained in the well and the weaker the signal.
Advantage
In competitive ELISA, antigens do not need purification before measurement. The process has also a higher specificity than indirect ELISA.
The ELISPOT assay
The enzyme linked immunospot (ELISPOT) assay is a modification of the ELISA assay, which allows the quantitative determination of the number of cells in a population that are specific for a given antigen or an antigen for which one has a specific antibody. Like sandwich ELISA, the ELISPOT assay derives its specificity and sensitivity by employing high affinity capture and detection antibodies and enzyme-amplification. The procedure for the ELISPOT assay is given below.
Procedure
- The microtiter plate wells are coated with the capture antigen or capture antibody. Capture antigen is recognized by the antibody of interest, and capture antibody is specific for the antigen whose production is being assayed.
- ELISPOT assay is commonly used to detect cytokine secreted from different cells.
- A cell suspension with some members secreting cytokine is added to the wells coated with relevant antibodies (capture antibodies). It is incubated for a certain period (figure 4).
- The wells are washed after the incubation period, and enzyme-labeled anti-cytokine antibodies (detection antibodies) are added.
- In the next step, the wells are re-washed to remove any unbound antibodies. After washing the wells, a chromogenic substrate that forms an insoluble colored product is added. The substrate forms an insoluble colored product.
- The colored product precipitates and forms a spot only on the areas of the wells where cytokine-secreting cells had been deposited.
- The number of colored spots determines the number of cytokine-secreting cells present in the added cell suspension.
Advantages
The elispot assay has higher sensitivity than other assays. This assay can make use of frozen or thawed biological samples. This method requires minimum biological samples and is also compatible with other assays.
CONCLUSION
The Interaction between antigen and antibody is a bimolecular association, which does not lead to an irreversible chemical alteration in either the antibody or the antigen. The ELISA is a type of antigen-antibody interaction. It is an immunological assay first described by Eva Engvall and Peter Perlmann in 1971. Various enzymes, such as alkaline phosphatase, horseradish peroxidase, and β-galactosidase, have been used for ELISA.
There are four types of ELISA methods, e.g., direct ELISA, indirect ELISA, sandwich ELISA, and competitive ELISA. Direct ELISA is the simplest type of ELISA. This method is used to detect and measure antigen concentration in a sample. Indirect ELISA is the method of choice to detect the presence of serum antibodies against human immunodeficiency virus (HIV), the causative agent of AIDS. Sandwich ELISA measures the antigen between two layers of antibodies. The two layers of antibody consist of capture and detection antibody. Competitive ELISA helps to measure the concentration of antigens. The enzyme-linked immunospot (ELISPOT) assay is a modification of the ELISA assay, which allows the quantitative determination of the number of cells in a population that are specific for a given antigen or an antigen for which one has a specific antibody.
You may also like:
- The antigen-antibody interaction: Agglutination reaction
- The antigen-antibody interaction: Precipitation reaction

I, Swagatika Sahu (author of this website), have done my master’s in Biotechnology. I have around fourteen years of experience in writing and believe that writing is a great way to share knowledge. I hope the articles on the website will help users in enhancing their intellect in Biotechnology.