


In this article, I describe the genome and the complexity within it.
Genome and Genome Annotation
The genome contains all the essential information an organism requires for its growth and functioning. It represents the entire collection of genetic material within an organism. A genome sequence comprises a lengthy string of the nucleotides A, G, T, and C, all of which must be accurately identified. Genome annotation is analyzing this sequence to determine the locations, functions, and other important features of genes. This step is crucial for transforming raw sequence data into meaningful information that researchers can use.
Using web-based computational tools, scientists can identify gene locations and suggest possible functions by comparing them to genes that have already been studied in other genomes. One widely used tool for this is BLAST (Basic Local Alignment Search Tool), which quickly scans genome databases for sequences similar to the one under investigation. This makes it particularly useful for exploring the potential function of a specific gene.

In every newly sequenced genome, a significant portion—around 40%—remains uncharacterized, presenting a major challenge for researchers. Determining the functions of these unknown genes is likely to take many years. Currently, much of the research is centered on protein-coding genes. By disabling a gene and observing changes in an organism’s growth or other traits, scientists can gain insights into the role of the protein the gene encodes. For some organisms, such as Saccharomyces cerevisiae and the plant Arabidopsis thaliana, researchers have created gene knockout libraries using genetic engineering to aid in this process.
The Various Sequences in the Human Genome
In some respects, humans are not as genetically complex as once believed. Earlier estimates suggested that the human genome, which spans approximately 3.2×109 base pairs, contained around 100,000 genes. However, recent discoveries have shown that we possess only about 20,000 protein-coding genes. That’s fewer than rice, which has about 38,000 genes, and only slightly more than a nematode worm (19,700 genes) or a fruit fly (13,600 genes).
Introns and Exons
In other respects, humans and eukaryotes are more complex than we once thought. Many eukaryotic genes include one or more stretches of DNA that do not directly contribute to the amino acid sequence of the resulting protein. These non-coding regions disrupt the otherwise straightforward connection between a gene’s DNA sequence and the sequence of the protein it produces. These interruptions are known as introns (intervening sequences), while the portions that code for proteins are called exons (Figure 1). In contrast, introns are rare in bacterial genes.
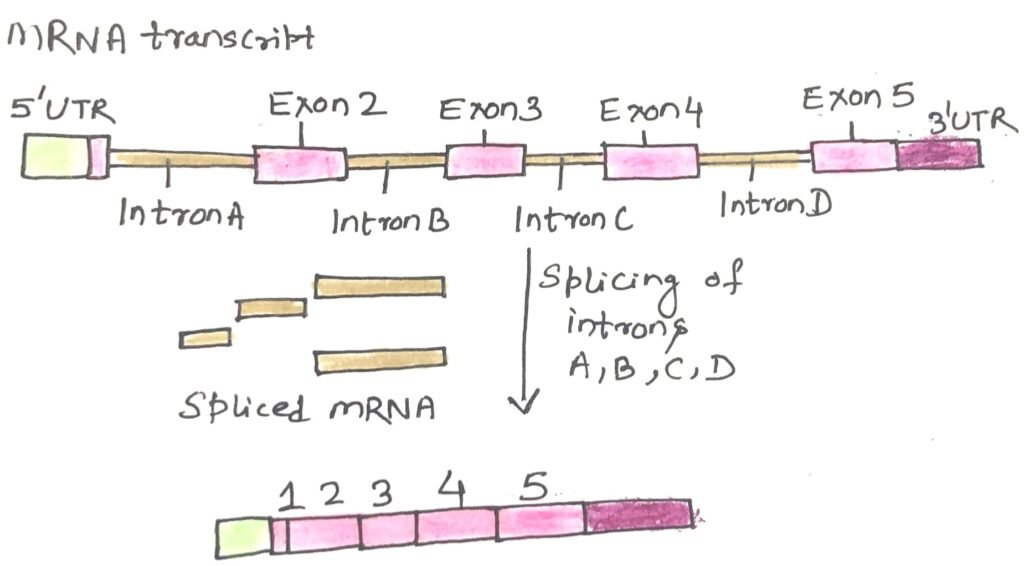
Introns are removed from the initial RNA transcript through splicing, resulting in a mature RNA strand that can be translated into a continuous protein sequence. Often, each exon corresponds to a specific functional domain within a larger, multi-domain protein. While humans share many of these protein domains with organisms like plants, worms, and flies, the way these domains are combined is more intricate in humans, contributing to a greater diversity of proteins in our proteome.
Additionally, variations in gene expression and RNA splicing allow for different combinations of exons to be assembled, meaning that a single gene can give rise to multiple distinct proteins. This phenomenon, known as alternative splicing, is significantly more prevalent in humans and other vertebrates than in simpler organisms like worms or bacteria, enabling the generation of a broader and more complex range of proteins.
Eukaryotic Genes Contain More Intron DNA
In mammals and many other eukaryotes, genes generally contain far more intron DNA than exon DNA. While the exact roles of introns remain largely unknown, they make up a significant portion of gene structure. Less than 1.5% of human DNA directly codes for proteins—this is the exon portion. However, when introns are factored in, genes that encode proteins account for up to 30% of the human genome.
Transposons are Mobile Elements in the Human Genome
A large portion of the human genome consists of non-coding DNA, much of which consists of repetitive sequences originating from transposons. These are segments of DNA, typically spanning a few hundred to several thousand base pairs, that have the unique ability to shift positions within the genome. First discovered in corn by Barbara McClintock, who referred to them as transposable elements, transposons are essentially molecular parasites that inhabit the genomes of nearly all organisms. The human genome contains various types of transposons, most of which are purely DNA-based. However, some, known as retrotransposons, resemble retroviruses in function. These elements move through the genome via RNA intermediates, which are then converted back into DNA through the process of reverse transcription.
In the human genome, a few transposons remain active and can still move, though infrequently. However, the majority are inactive, mutated remnants from earlier stages of evolution. The movement of transposons can also cause the rearrangement of other DNA sequences, a process that has significantly influenced the course of human evolution.
The ENCODE Project Decodes the Dark Matter of the Genome
After accounting for protein-coding genes and transposons, roughly 25% of the human genome remains. To better understand this portion, the ENCODE (Encyclopedia of DNA Elements) project was launched in 2003 by the U.S. National Human Genome Research Institute as a follow-up to the Human Genome Project. Through the efforts of a global network of research teams, ENCODE has shown that most of the human genome is either transcribed into RNA in at least one cell or tissue type or plays a role in chromatin organization. A significant part of the remaining 20% of non-coding DNA includes regulatory elements that influence the activity of the approximately 20,000 protein-coding genes, as well as many genes that produce functional RNAs. Many mutations associated with human genetic diseases are part of this non-coding DNA. It may affect the regulation of one or more genes.
SSRs and STRs
Approximately 3% of the human genome is made up of highly repetitive sequences known as simple sequence repeats (SSRs). These sequences are typically under 10 base pairs in length and can be repeated millions of times within a cell, arranged in short stretches of tandem repeats. Notable examples of SSR DNA are located in centromeres and telomeres. For instance, human telomeres can contain up to 2000 consecutive copies of the GGTTAG sequence. In addition to these, shorter simple sequence repeats—referred to as short tandem repeats (STRs)—are scattered throughout the genome. These segments usually consist of a few dozen tandem repeats and are commonly used in forensic DNA analysis.
Genetic variations
The human population contains millions of single-base differences known as single-nucleotide polymorphisms, or SNPs. On average, one out of every 1000 base pairs differs between individuals. A significant portion of these variations are SNPs. Beyond these, people also exhibit a broad spectrum of larger genetic changes, including deletions, insertions, and minor rearrangements. These often subtle genetic variations are responsible for the diversity we observe among humans, such as variations in hair color, height, foot size, vision, sensitivity to medications, and even behavior.
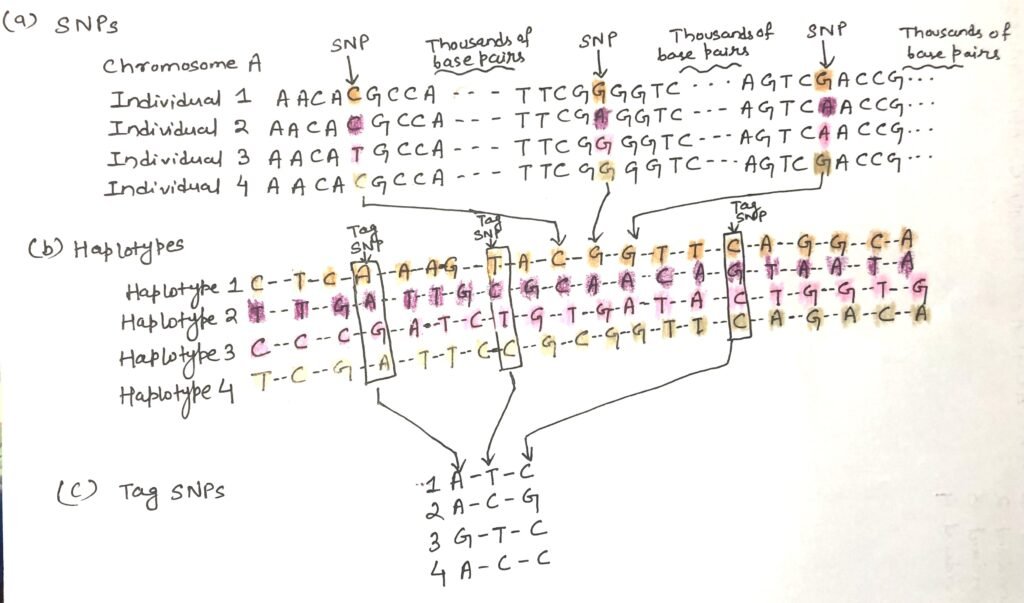
During meiosis, genetic recombination shuffles small genetic variations, resulting in different combinations of genes being passed down. However, when genetic variations like SNPs are located close together on a chromosome, they tend to remain linked and are typically inherited as a unit because recombination seldom separates them. This cluster of closely associated SNPs is called a haplotype. Haplotypes serve as useful genetic markers for identifying specific human populations and individuals within those groups.
Haplotype Identification and Its Use in Tracing Human Ancestry
Identifying a haplotype involves a series of steps. Initially, scientists examine genomic DNA from multiple individuals (Figure 2a) to pinpoint positions where single-nucleotide polymorphisms (SNPs) occur in the human population. Although SNPs within a potential haplotype may be separated by thousands of base pairs, they are still considered “close” in the context of chromosomes that span millions of base pairs. Next, a group of SNPs that are commonly inherited together is defined as a haplotype (Figure 2b).
Each haplotype is characterized by the specific nucleotide bases found at each SNP position within the group. To efficiently distinguish haplotypes, researchers identify a smaller subset of these SNPs—known as tag SNPs—which can uniquely represent the full haplotype (Figure 2c). By sequencing only these tag SNPs, it becomes easier to determine which haplotypes are present in an individual’s DNA. Some haplotypes, particularly those in the mitochondrial DNA and the Y chromosome, are remarkably stable. These can be used as genetic markers to trace patterns of human migration across generations.
Conclusion
The genome contains all the genetic information needed for an organism’s growth and function. It is represented by a sequence of A, G, T, and C nucleotides. Genome annotation helps identify gene locations and functions using tools like BLAST, which compares unknown sequences to known ones. Despite this, about 40% of genes in new genomes remain uncharacterized. Research mainly focuses on protein-coding genes. It often uses gene knockouts in model organisms like Saccharomyces cerevisiae and Arabidopsis thaliana to study gene functions.
Splicing removes introns from RNA, producing mature RNA that codes for proteins. Exons often align with functional protein domains, and humans combine these domains more complexly than simpler organisms. Alternative splicing—more common in humans—allows one gene to produce multiple proteins. Though their function is unclear, introns make up most of gene structure. While only about 1.5% of human DNA codes for proteins, including introns, protein-coding genes cover around 30% of the genome.

A large portion of the human genome consists of non-coding DNA. Much of this is made up of repetitive sequences originating from transposons. These are segments of DNA, typically spanning a few hundred to several thousand base pairs. These have the unique ability to shift positions within the genome.
Humans have millions of single-base differences called SNPs, occurring about once every 1000 base pairs. Along with larger changes like insertions or deletions, these variations drive human diversity in traits and responses. During meiosis, recombination shuffles genes, but nearby SNPs often stay linked and are inherited together as haplotypes. These haplotypes help identify populations and individuals genetically.
You may also like:

I, Swagatika Sahu (author of this website), have done my master’s in Biotechnology. I have around fourteen years of experience in writing and believe that writing is a great way to share knowledge. I hope the articles on the website will help users in enhancing their intellect in Biotechnology.