
This article describes the evolutionary origin of mitochondrial genes and examines how mutations in mitochondrial DNA influence cellular function, aging, and human disease. Mitochondria are essential organelles responsible for generating most of the energy required for cellular activities. Unlike other cellular components, they possess their own genetic material, reflecting a unique evolutionary history linked to ancient bacteria. Over time, mitochondrial genes have become closely integrated with the nuclear genome to support efficient cellular metabolism.
Overview of the Mitochondrial Genome
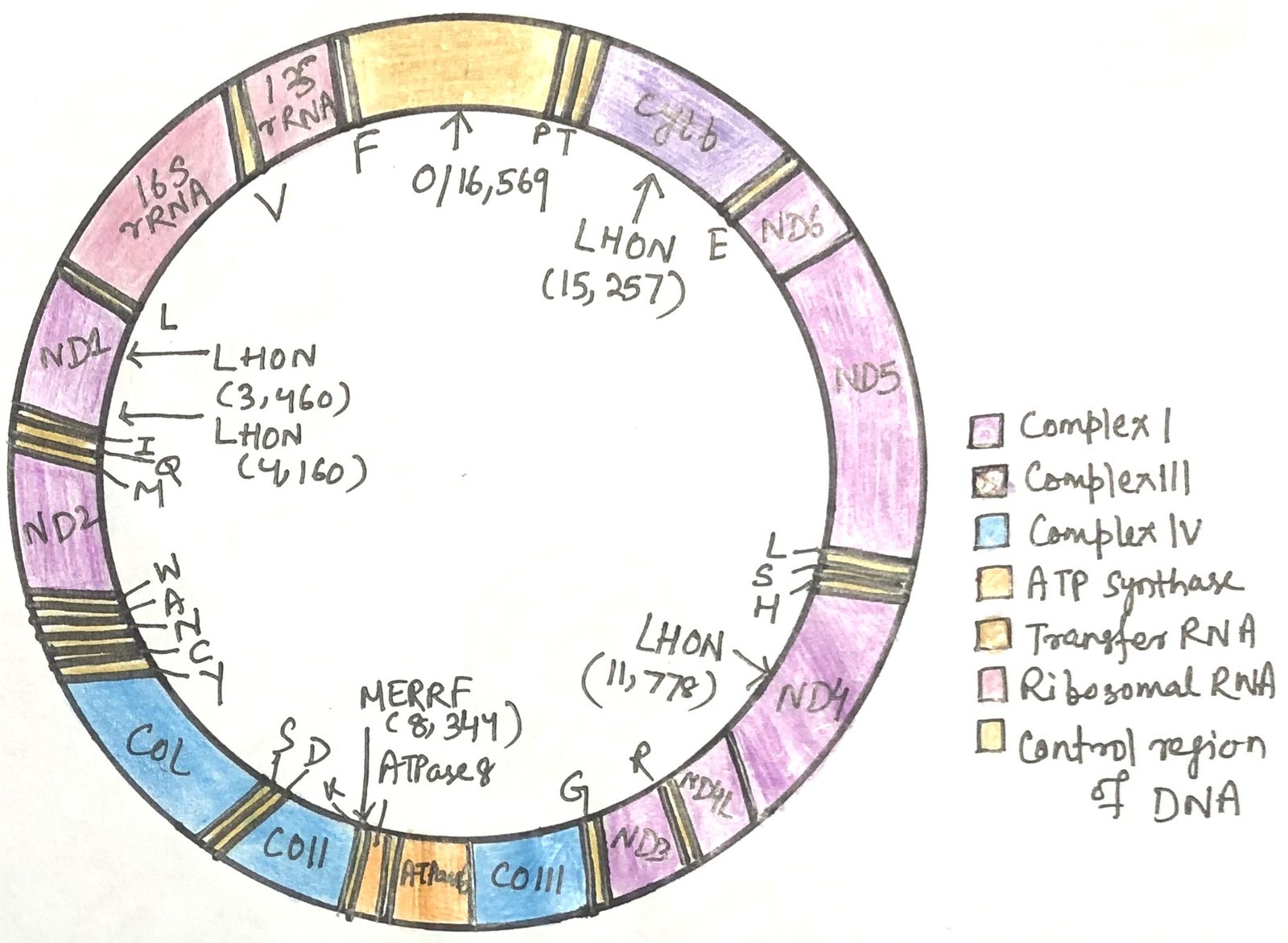
Mitochondria possess a small, independent genome made of circular, double-stranded DNA. A single cell usually contains hundreds to thousands of mitochondria. Each organelle carries several copies of this genome, typically around five. In humans, mitochondrial DNA spans 16,569 base pairs and contains 37 genes (Figure 1). Thirteen of these genes produce protein components of the respiratory chain, while the rest encode rRNA and tRNA molecules needed for mitochondrial protein synthesis. Mitochondria therefore have their own specialized ribosomes, which differ from those in the cytoplasm. Despite this autonomy, most mitochondrial proteins, approximately 1,100 types, are encoded by nuclear DNA. They are produced on cytoplasmic ribosomes and later transported into mitochondria, where they are assembled and function.
Endosymbiotic Origin of Mitochondria
The presence of mitochondrial DNA, ribosomes, and transfer RNAs provides strong evidence for the endosymbiotic theory, which proposes that mitochondria originated from ancient aerobic bacteria. According to this idea, early organisms capable of using oxygen for respiration and ATP generation were free-living bacteria. Anaerobic primitive eukaryotic cells later gained the ability to perform oxidative phosphorylation by forming a symbiotic partnership with such bacteria residing within their cytoplasm. Over long evolutionary periods, many bacterial genes were transferred to the host cell nucleus, and the once-independent bacteria gradually evolved into modern mitochondria.
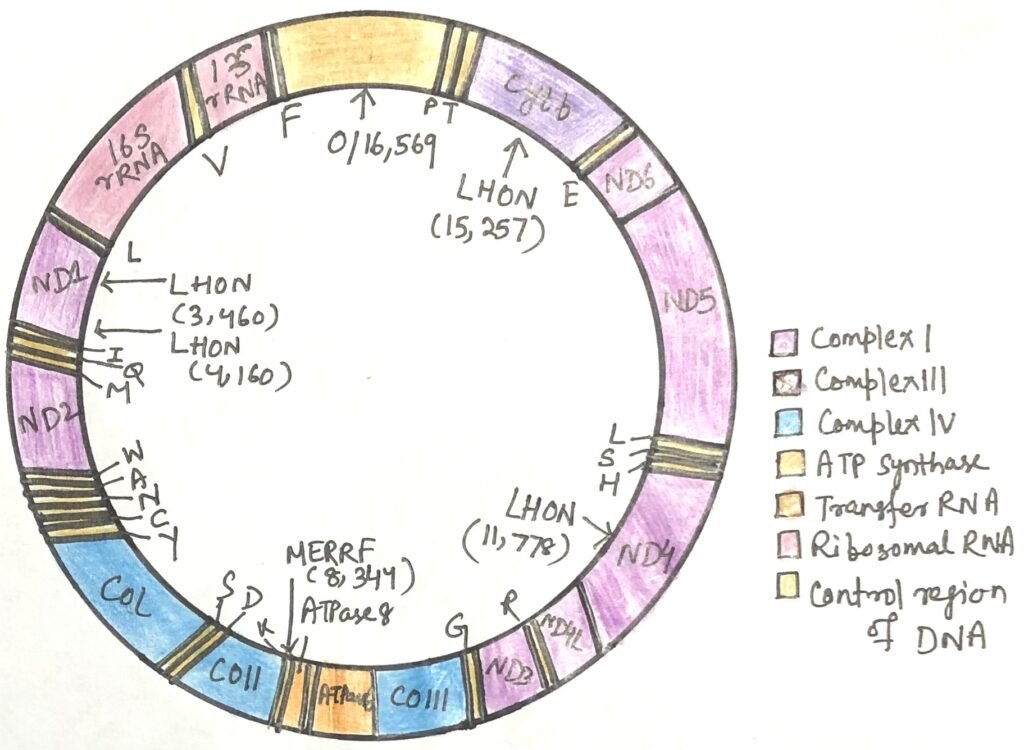
This hypothesis assumes that ancestral bacteria already possessed the enzymes required for oxidative phosphorylation and predicts that present-day bacterial descendants should exhibit respiratory systems similar to those of eukaryotic cells. Aerobic bacteria perform NAD-linked electron transfer from metabolic substrates to oxygen while simultaneously generating ATP from ADP. In these organisms, dehydrogenase enzymes operate in the cytosol, whereas the respiratory chain is embedded in the plasma membrane. During electron transport to oxygen, carrier proteins pump protons across the membrane, creating a gradient. In bacteria such as E. coli, FoF₁ ATP synthase complexes are located in the plasma membrane; the F₁ portion extends into the cytosol and produces ATP as protons flow back into the cell through the Fo channel.
Chemiosmosis and Early Bioenergetics
Proton pumping during bacterial respiration does more than support ATP production. The proton gradient formed across the plasma membrane also powers several other essential cellular activities. Many bacterial transport systems use this gradient to import nutrients from the environment against their concentration gradient by coupling nutrient uptake with the inward movement of protons. In addition, bacterial flagella rotate through the action of proton-driven molecular motors, often described as “proton turbines.” These rotary systems are not powered by ATP directly but instead rely on the electrochemical potential created by respiration-linked proton movement across the membrane (Figure 2). Such observations suggest that the chemiosmotic mechanism is an ancient energy-conserving strategy that likely evolved well before the appearance of eukaryotic cells.
Maternal Inheritance and Heteroplasmy in Mitochondrial Disorders
A distinctive aspect of mitochondrial genetics is the wide variation in how mitochondrial DNA (mtDNA) mutations affect different cells and individuals. Each cell contains hundreds or even thousands of mitochondria, and every mitochondrion carries multiple copies of its genome. In animals, mitochondria are inherited almost exclusively from the mother. This occurs because egg cells are large and contain about 10⁵–10⁶ mitochondria, whereas sperm cells are much smaller and carry only a few hundred to a thousand. In addition, shortly after fertilization, specialized maternal cellular structures actively eliminate mitochondria contributed by the sperm by engulfing and degrading them.
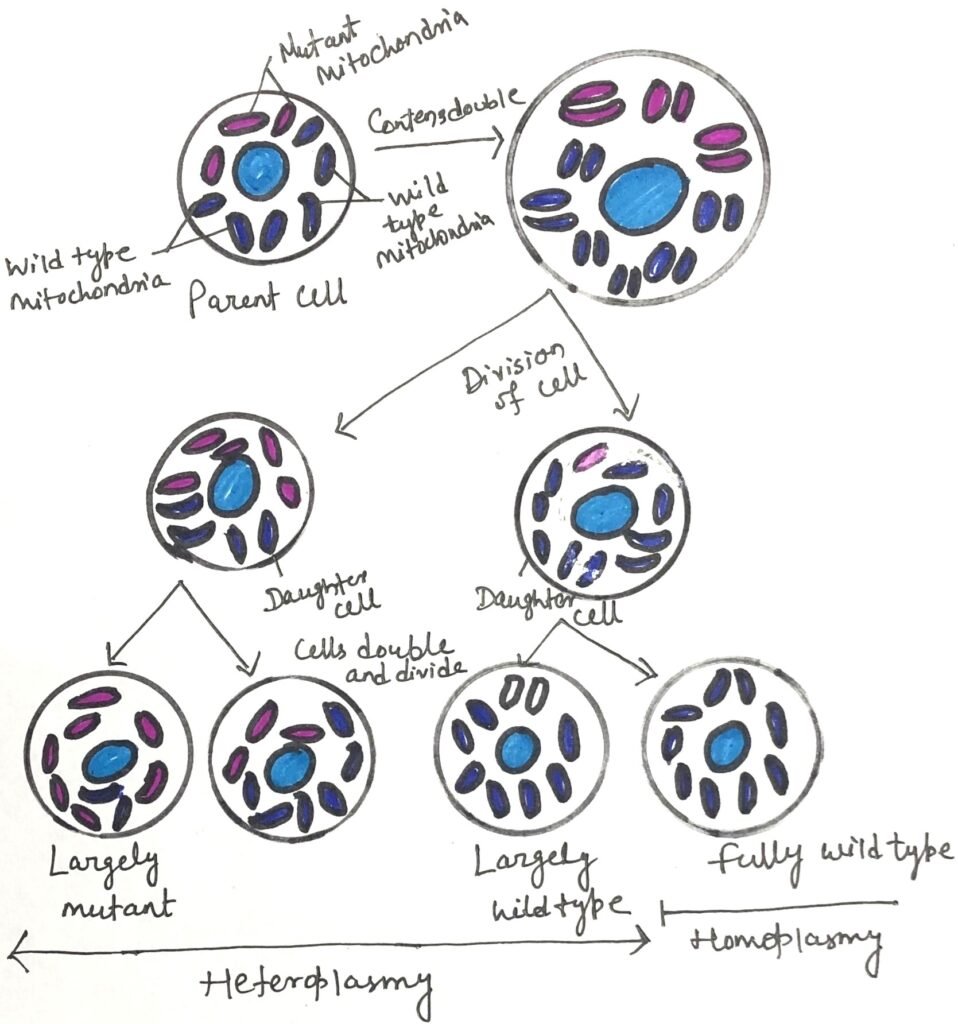
Let us consider a situation where a mutation arises in the mitochondrial genome of a germ cell that will give rise to oocytes. At first, most mitochondria in that cell remain normal, with only a single mitochondrion carrying the mutation. As the germ cell divides during oocyte development, the defective mitochondrion replicates and randomly distributes its copies among the daughter cells. Over time, different developing egg cells end up with varying proportions of mutant mitochondria (Figure 3).
After fertilization and throughout embryonic development, this uneven distribution continues as cells divide, producing tissues that contain different levels of mutated mitochondria. This condition, known as heteroplasmy, leads to a wide spectrum of outcomes. Cells dominated by normal mitochondria typically function normally, while those containing higher proportions of defective mitochondria may show impaired function. When a mutation causes disease, the distribution and proportion of affected mitochondria determine how severely individuals with the same mtDNA mutation experience symptoms.
Prevalence and Tissue Sensitivity to Mitochondrial Mutations
Approximately one in every 5,000 individuals carries a mutation in a mitochondrial protein that limits the cell’s ability to generate ATP. Cells with especially high energy demands, such as neurons, skeletal and cardiac muscle cells, and pancreatic β-cells, are particularly vulnerable to reduced ATP production. Because these tissues rely heavily on oxidative phosphorylation, mitochondrial dysfunction affects them earlier and more severely than other tissues.
A group of inherited disorders known as mitochondrial encephalomyopathies primarily affects the brain and skeletal muscles. These pass from mothers to their offspring. One well-known example is Leber hereditary optic neuropathy (LHON), a rare condition that damages the central nervous system, especially the optic nerves, leading to loss of vision in early adulthood. In many cases, LHON arises from a single nucleotide change in the mitochondrial ND4 gene that substitutes histidine for arginine in a subunit of Complex I.
This alteration weakens electron transfer from NADH to ubiquinone, reducing ATP production. Although mitochondria can still generate limited ATP through electron transfer from succinate, this supply is insufficient for the high metabolic demands of neurons, including those in the optic nerve. Another single-base mutation in the mitochondrial cytochrome b gene of Complex III can also cause LHON, indicating that the disease stems from an overall decline in mitochondrial performance rather than a defect confined to Complex I.
Major Mitochondrial Disorders and Nuclear Gene Involvement
Myoclonic epilepsy with ragged-red fibers (MERRF) results from a mutation in a mitochondrial gene encoding a lysine-specific tRNA. This defect disrupts the synthesis of several mitochondrial proteins, leading to symptoms such as involuntary muscle jerking. In affected individuals, skeletal muscle fibers often contain abnormally shaped mitochondria that may display distinctive paracrystalline inclusions. Other mutations in mitochondrial genes cause progressive muscle weakness in mitochondrial myopathy and lead to the enlargement and deterioration of the heart muscle in hypertrophic cardiomyopathy.
Mitochondrial diseases are not limited to mutations within mitochondrial DNA itself. They can also arise from changes in any of the roughly 1,100 nuclear genes that encode mitochondrial proteins. For instance, mutations in the nuclear gene coding for the Complex IV protein COX6B1 can lead to severe abnormalities in brain development, along with thickening of the heart muscle walls.
Conclusion
Mitochondria represent a remarkable link between ancient bacterial life and modern eukaryotic cells. Their independent genome, bacterial-like bioenergetic systems, and maternal inheritance all support the idea that they originated from an ancient symbiotic partnership that transformed cellular evolution. Over time, the transfer of many mitochondrial genes to the nuclear genome created a highly integrated system in which both genomes cooperate to sustain cellular energy production.
At the same time, the unique features of mitochondrial DNA make it especially vulnerable to mutation. Continuous exposure to reactive oxygen species, limited repair mechanisms, and the phenomenon of heteroplasmy contribute to the gradual accumulation of mitochondrial defects. These changes play an important role in aging and underlie a wide range of inherited and acquired diseases, particularly in tissues with high energy demands.
Understanding the origin, function, and mutation of mitochondrial genes, therefore, provides valuable insight into evolution, human health, and disease. Ongoing research into mitochondrial biology continues to open new possibilities for diagnosing, preventing, and treating disorders linked to impaired cellular energy metabolism.
You may also like:

I, Swagatika Sahu (author of this website), have done my master’s in Biotechnology. I have around fourteen years of experience in writing and believe that writing is a great way to share knowledge. I hope the articles on the website will help users in enhancing their intellect in Biotechnology.